

研究成果简介
氮化物因其不同寻常的物理化学特性而备受关注,例如从可见光到紫外光区域的宽带隙、超导特性、高能量密度、和超高硬度以及较好的化学和热稳定性。受到石墨烯的启发,超薄二维氮化物由于在二维尺度下呈现出的新的物理化学特性也引起了科学家极大的兴趣,例如,与体材料纤锌矿型GaN相比,实验发现二维GaN具有更大的带隙、更高的泊松比、蓝移的光致发光发射谱以及更高的内量子效率。IV族氮化物是一类重要的氮化物,Ge3N4、Si3N4和g-C3N4被广泛应用于光催化、高温陶瓷、微电子和光电子元件、机械、航空航天等领域。文献调研发现,在二维尺度下,二维C-N化合物出现多种新的化学计量比(C3N、C2N、C4N3、CN、C3N4以及C2N3),而鲜有研究涉及二维A-N体系(A=Sn/Ge/Si)。同时,二维尺度下多氮结构的存在形式和稳定性机制也是一个值得深入探究的课题。
鉴于此,在王俊杰和Gilles Frapper两位教授的指导下,博士生张恒利用第一性原理进化算法(USPEX+VASP)开展了二维IV族氮化物的结构搜索和电子性质研究。通过详细的二维定成分和变成分结构搜索,确定了10种二维IV族氮化物原型结构,包括C2/m A4N、P3m1 A3N、P3m1 A2N、P3m1 A3N2、P6m2 AN、P3m1 AN、P62m A3N4、P3m1 A2N3、P421m AN2和P3m1 AN3(参见图1)。首先,基于四个稳定性标准(热力学稳定性、弹性稳定性、动力学稳定性和热稳定性),我们从30个结构中确定了25个具有较高实验合成可行性的二维结构。其次,详细分析讨论了该10个二维结构原型的结构特征和可能的电子性质,并利用DFT计算验证了以上的谈论。HSE06杂化泛函计算表明,二维P3m1 A3N、P6m2 AN、P3m1 AN、P62m A3N4、P421m AN2和P3m1 AN3均为半导体材料,而超配位(非局域)的A原子导致C2/m A4N、P3m1 A2N和P3m1 A3N2表现出金属导电性(图2-3)。进一步,我们结合电荷局域密度函数(ELF)、晶体轨道哈密顿布居(COHP)、键级(BO)以及分子轨道理论解释了以上原型结构的半导体性或者金属性导电特性。同时,考虑电子自旋效应,二维P3m1 A2N3为反铁磁半导体,反铁磁态比铁磁和无磁态能量高400~800 meV/f.u。与二维In2S3(24个价电子,本征半导体)相比,二维P3m1 A2N3(23个价电子)的铁磁态来源于体系N原子的未配对电子。HSE06杂化泛函计算表明二维P3m1 Sn2N3、Ge2N3和Si2N3的带隙分别为0.677、1.285和2.321 eV(图4)。最后,利用GGA-PBE泛函研究了所有体系的弹性性质,发现二维P421m SiN2在全平面内具有拉胀效应,计算的负泊松比为ν12=ν21=-0.146(图5)。分析表明,二维P421m SiN2中四面体SiN4特殊的空间取向和应变下Si-to-N2的电荷转移共同导致了体系的负泊松比效应。
以上研究工作近期以题目“Prediction of Two-dimensional Group IV-nitrides AxNy (A=Sn/Ge/Si): Diverse Stoichiometric Ratios, Ferromagnetism, and Auxetic Mechanical Property”发表在Journal of Physical Chemistry Letters上(J. Phys. Chem. Lett. 2022, 13, 9316-9325)。该研究得到了国家自然基金(No. 51872242和No. 52111530033)和中央高校科研基金(D5000200142)的支持,博士生张恒受到了基金委2019中法蔡元培项目的支持。该论文的通信作者是王俊杰教授和Gilles Frapper教授,西北工业大学凝固技术国家重点实验室为论文第一完成单位。
图文导读
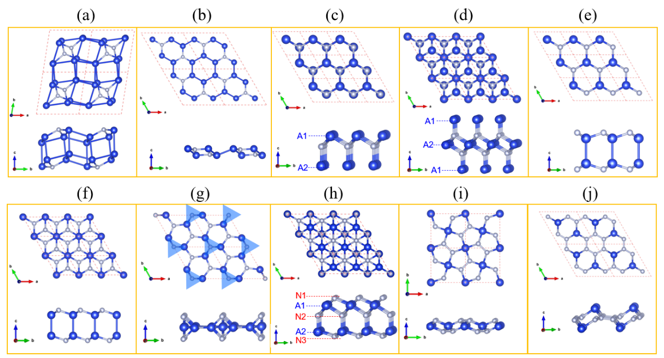
Figure 1. Ten 2D prototypes of predicted 2D group IV-nitrides AxNy: (a) C2/m A4N, (b) P3m1 A3N, (c) P3m1 A2N, (d) P3m1 A3N2, (e) P6m2 AN, (f) P3m1 AN, (g) P62m A3N4, (h) P3m1 A2N3, (i) P421m AN2, and (j) P3m1 AN3. In (a-j), blue and silver balls indicate A (Sn/Ge/Si) and N atoms, respectively. In (g), A3N2 trigonal bipyramids are indicated using shadow blue triangles.
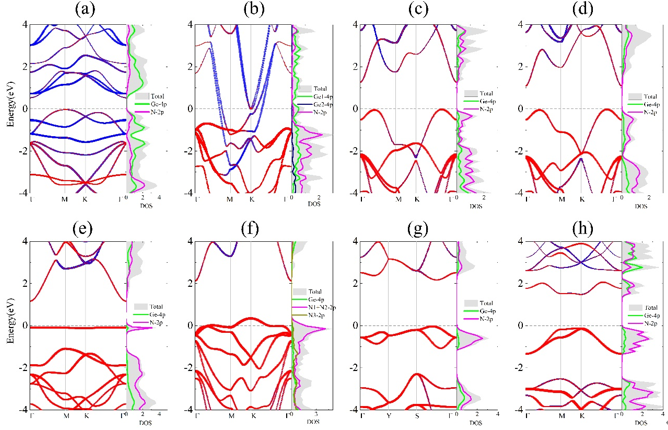
Figure 2. Projected electronic band structures and density of states (DOS) (GGA-PBE level of theory) of 2D GexNy: (a) P3m1 Ge3N, (b) P3m1 Ge3N2, (c) P6m2 GeN, (d) P3m1 GeN, (e) P62m Ge3N4, (f) P3m1 Ge2N3, (g) P421m GeN2, and (h) P3m1 GeN3. In the projected band structures, blue and red colors indicate the energy states contributed by Ge and N atoms, respectively. In (a-h), the Fermi level is set as zero.
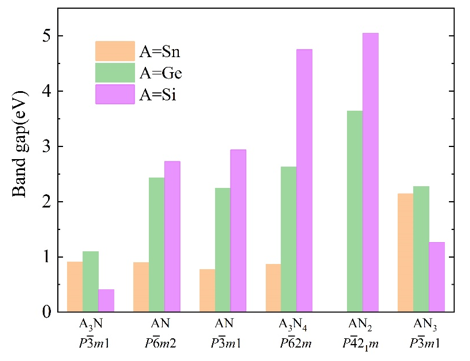
Figure 3 Calculated band gaps of six semiconducting 2D AxNy protypes at the HSE06 level of theory.
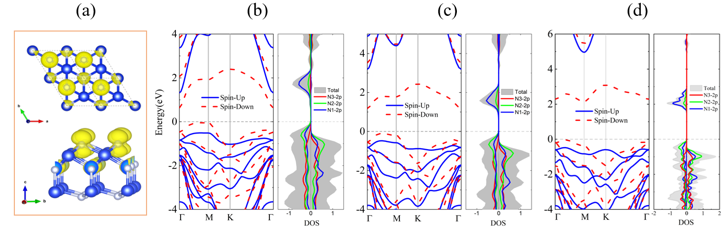
Figure 4. Ferromagnetic properties of 2D P3m1 A2N3: (a) spin charge distribution of the ferromagnetic state; spin-polarized band structures and DOS of (b) 2D P3m1 Sn2N3, (c) 2D P3m1 Ge2N3, and (d) 2D P3m1 Si2N3 at HSE06 level.
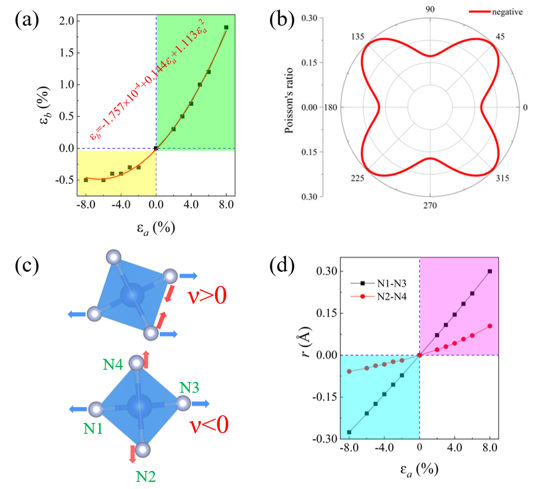
Figure 5. Negative Poisson’s ratio of 2D P421m SiN2: (a) The strain response along b direction (εb) versus the strain along a direction (εa); (b) In-plane Poisson’s ratio as a function of the angle θ; (c) Deformation of SiN4 tetrahedra under the external strain; (d) The distance variation (r) of N1-N3, N2-N4 pairs versus εa in the SiN4 tetrahedra. The blue and red arrows in (c) indicate the distance expand and contract, respectively.
文章链接:https://doi.org/10.1021/acs.jpclett.2c02376