

研究成果简介:
在电子化合物的发现过程中,大量富电子的非电子化合物被报道。这一类材料具有成为电子化合物的潜力,但未得到大家的关注,迄今为止,几乎没有工作以这些材料为着力点,进行改性获取新型电子化合物。本研究证明,在富电子的非电子化合物进行适当的掺杂替换,可以增强材料的电子化合物特征,间隙电子与最近邻阳离子之间的弱的成键态是电子化合物形成的关键。与此同时,我们的工作指出,在多阳离子的材料中,由电负性差的阳离子构成笼原子是电子化合物形成的必要条件。本文以富电子的P63/mcm- Hf5Si3(Hf为+4价,Si为-4价,则每分子多了8个电子)为例,应用第一性原理计算,对其电子结构进行了系统的深入研究,通过元素掺杂置换,改变了非电子化合物材料 Hf5Si3间隙电子的能量,进而获得了前所未有的电子化合物材料Ca3Hf2Si3。在此基础上,我们在 29 种二元非电子化物材料中适当掺杂获得了34 种动态稳定的三元电子化物。该工作提供了新的方式去设计富电子型电子化合物,对于理解电子化合物的形成机理,进一步设计新型电子化合物具有深远影响。

背景介绍:
电子化合物是一类特殊的新型功能材料,聚集在结构内间隙位置的高能非核电子,对材料的性能产生巨大影响,如导致较低的功函数。已有的研究报道电子化合物在催化剂、磁性、超导等多个领域具有应用前景。目前稳定的电子化合物数目较少,发现新型电子化合物是电子化合物研究领域的长期热点,然而电子化合物的设计与发现受限于当前人们对于其形成机理的理解,电子化合物的发现主要依托于已有材料的筛选,在筛选的过程中,大量的富电子的非电子化合物的被报道,这些材料的改性工作至今未得到重视。
本文亮点:
近日,博士生李琨应用第一性原理计算,对P63/mcm-Hf5Si3的电子结构进行了系统的深入研究,发现缺电子的Hf5Si3在适当位置掺杂Ca原子可以得到为电子化合物。与以往研究中直接寻找富电子的电子化合物不同,本研究将重心放在对具电子化合物潜力的富电子化合物进行成分调节,结果表明具有较弱电负性的阳离子取代电负性大的阳离子作为间隙电子的最近邻原子,可以有效提高间隙电荷的能量,获得高能间隙电子,进而实现激活材料的电子化合物特征。相关研究成果以“Discovery of electrides in electron-rich non-electride materials via energy modification of interstitial electrons”为题发表在期刊Advanced Functional Materials上。
图文导读:
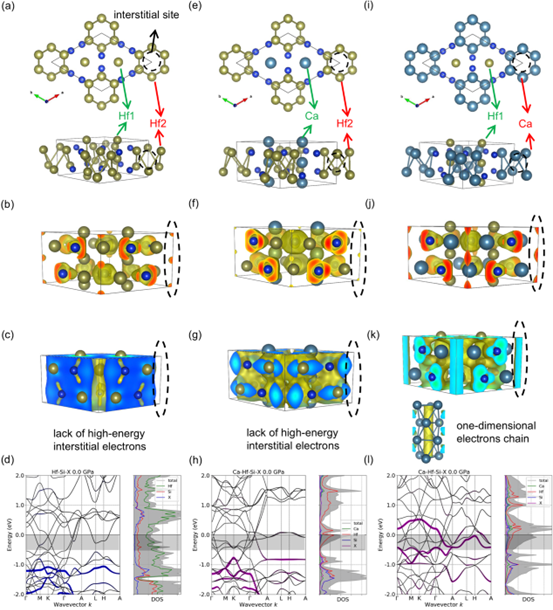
图 1. (a-d) Hf5Si3、(e-h) Ca2Hf3Si3 和 (i-l) Ca3Hf2Si3 的(a、e 和 i)晶体结构、(b、f 和 j)计算的电荷局域函数、(c、g 和 k)部分电荷密度(-0.5 < E-EFermi < 0 eV)和(d、h、 l) 计算的投影能带结构和态密度。 在 (b)、(f) 和 (j) 中电荷局域函数的等值面分别以 0.7、0.65 和 0.75 的值绘制。 (c)、(g) 和 (k) 中部分电荷密度的等值面分别以 0.0005、0.001 和 0.003 e/Å3 的值绘制
图1中(a-d)显示 Hf5Si3不是电子化合物, Hf5Si3的间隙位置虽然有局域电荷(图1b),但是能量较低(图1c和d),投影能带分析显示该部分电荷占据原理费米能级的能带。使用Ca原子取代Hf1得到Ca2Hf3Si3,进一步的电子结构分析显示,间隙电荷能量仍很低(图1f-h),表明该结构不是电子化合物。而使用Ca取代Hf2得到Ca3Hf2Si3,部分电荷密度(-0.5 < E-EFermi < 0 eV)显示高能态电子分布在Ca3Hf2Si3的间隙位置,形成一维电子链(图1k),投影能带分析显示该部分电荷占据高能能带,穿越费米能级(图1f),上述现象表明该结构是电子化合物。

图2. (a) Ca3Hf2Si3和(b) Ca2Hf3Si3投影态密度图。(c) Hf-X (Hf5Si3), Hf-X (Ca2Hf3Si3), Ca-X (Ca3Hf2Si3), (d) C-C (diamond), Fe-Fe (Fe), Na-Cl (NaCl), (e) Y-X (Y5Si3) 和 Ca-X (C12A7)晶体轨道哈密顿分布(COHP)
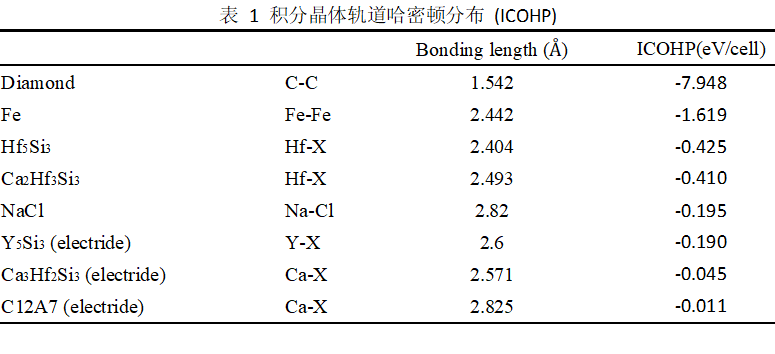
图2(a和b)显示Ca3Hf2Si3和Ca2Hf3Si3的差异主要来自于间隙电荷和最紧邻阳离子的耦合作用。在Ca3Hf2Si3中间隙电子X轨道和Ca-d轨道主要在高能级-0.7~0 eV区间重叠,而Ca2Hf3Si3中间隙电子X轨道和Hf-d轨道主要在低能级-1.5~-0.7 eV区间重叠。晶体轨道哈密顿分布(c)进一步显示,Ca2Hf3Si3中间隙电子和Hf原子间具有更多的成键态(ICOHP=-0.41 eV/cell),而Ca3Hf2Si3中间隙电子和Ca原子成键态较弱(ICOHP=-0.045 eV/cell)。作为对比,我们也计算了常见的共价键、金属键、离子键以及电子化合物中间隙电子与最紧邻阳离子之间的COHP(c-e)和ICOHP(表 1),结果显示电子化合物中间隙电荷和最紧邻阳离子之间成键最最弱,只表现出离子键。

图3.Ca3Hf2Si3电子化合物的形成机理
基于此,考略到Ca(1.0)和Hf(1.3)之间的电负性的差异,我们对Ca3Hf2Si3和Ca2Hf3Si3的差异给出解释,并绘制示意图3。当Ca原子取代Hf2时,Ca低的电负性,降低对间隙电子的吸引力,成键态减弱,间隙电子能量升高,材料转变电子化合物。而当Ca取代Hf1是,Hf2隔绝了Ca对间隙电子影响,间隙电子的能量变化不大。
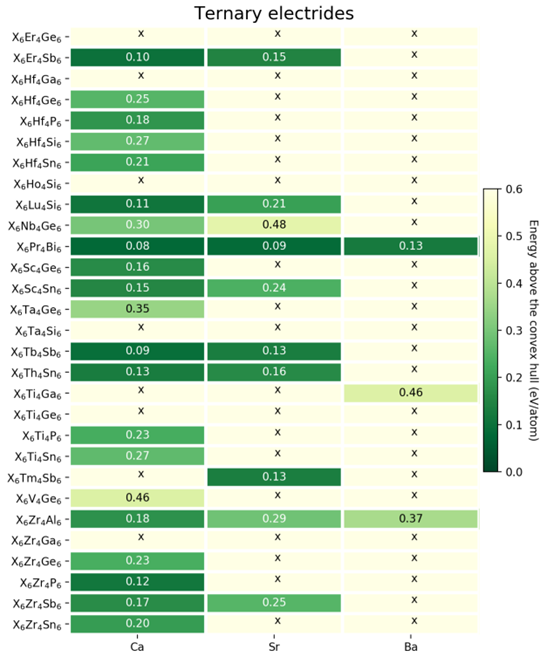
图4. 预测的三元电子化合物。 “×” 表示该结构动力学不稳定,数字表示该结构距离相图凸包线的距离
以Hf5Si3改性结果为基础,我们对29种富电子的非电子化合物进行改性得到34中动力学稳定的三元电子化合物,展示在图4。